Zespół Treachera Collinsa

Dyzostoza twarzowo-żuchwowa, znana bardziej jako zespół Treachera Collinsa, to rzadko występująca, uwarunkowana genetycznie choroba dotycząca zaburzeń rozwojowych w obrębie kości i innych tkanek twarzy. Cechami wyróżniającymi ten zespół od innych, są wyraźne cechy dysmorficzne, jak antymongoidalne ustawienie powiek (skośne oczy) i rozszczep powiek, niedorozwój kości jarzmowej, widoczny jako brak policzków, a także hipoplazja żuchwy - obserwowana pod postacią małej, cofniętej bródki. Charakterystyczny jest ponadto niedorozwój małżowin usznych i ucha wewnętrznego. Stwierdza się również rozszczep podniebienia i dysmorfie nosa. Rozwój intelektualny dziecka jest prawidłowy. Proponowane jest leczenie rekonstrukcyjne i plastyczne.
Synonimy:
- ang. Treacher Collins Syndrome, TCS - zespół Treacher-Collinsa,
- ang. Franceschetti-Zwahlen-Klein syndrome - zespół Franceschetti-Zwahlen-Klein,
- ang. mandibulofacial dysostosis, MFD - dyzostoza żuchwowo-twarzowa,
- ang. Treacher Collins-Franceschetti syndrome - zespół Treachera Collins'a-Franceschetti,
- ang. zygoauromandibular dysplasia - dysplazja jarzmowo-uszno-żuchwowo-twarzowa,
- ang. Mandibulofacial dysostosis without limb anomalies - dyzostoza żuchwowo-twarzowa bez wad kończyn.
Rozpowszechnienie: 1:50 000 żywych urodzeń.
Przyczyny i dziedziczenie
Wszystkie cechy zespołu wynikają ze wczesnych zaburzeń rozwoju zarodkowego pierwszego i drugiego łuku skrzelowego (łuki skrzelowe/gardłowe to struktury występujące w okresie embrionalnym, z których później wykształca się np. kosteczki słuchowe, błona bębenkowa, jama bębenkowa, trąbka słuchowa, jama sutkowa kości skroniowej, kość gnykowa, szczęka, żuchwa, gardło, migdałek podniebienny).
Mutacje są dziedziczone w sposób autosomalny dominujący lub recesywny. Zespół może być spowodowany mutacjami w genach:
- TCOF1 (81-93%% przypadków) i wówczas określany jest jako zespół Treachera Collinsa typu I,
- POLR1C - zespół Treachera Collinsa typu II,
- POLR1D - zespół Treachera Collinsa typu III (razem z II: dotyczą około 2% przypadków),
- u pozostałych pacjentów przyczyna genetyczna może być nieznana.
TCOF1 (ang. treacle ribosome biogenesis factor 1) znajduje się na ramieniu długim chromosomu 5, między pozycjami 32 i 33.1 (5q32-33.1) i koduje białko jąderkowe zwane treacle, które jest aktywne we wczesnych etapach rozwoju zarodkowego w strukturach przekształcających się w kości twarzy oraz inne jej tkanki. Mutacje w tym genie są odpowiedzialne za większość przypadków zespołu Treachera Collinsa. Są dziedziczone autosomalnie dominująco.
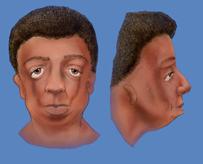 |
| Niedorozwój twarzy i dysmorfie http://www.primehealthchannel.com |
POLR1D (ang. RNA polymerase I subunit D) jest zlokalizowany na ramieniu długim chromosomu 13, w pozycji 12.2 (13q12.2), natomiast POLR1C (ang. RNA polymerase I subunit C) znajduje się na ramieniu krótkim chromosomu 6, w pozycji 21.1 (6p21.1). Obydwa geny zawierają instrukcje dla prawidłowego wytwarzania jednego z dwóch białek enzymatycznych: polimeraza I RNA i polimeraza III RNA, które uczestniczą w syntezie rybosomalnego RNA. Polimeraza III odpowiada także za wytwarzanie RNA transportującego. rRNA i tRNA łączą aminokwasy w białka funcjonalne, które są istotne dla naturalnej aktywności i przeżycia komórek, zwłaszcza tkanek twarzy. Mutacje w POLR1D są dziedziczone autosomalnie dominująco, a w POLR1C - autosomalnie recesywnie.
Podsumowując, mutacje TCOF1, POLR1D i POLR1C doprowadzają do obniżenia wytwarzania rRNA, co prawdopodobnie skutkuje do apoptozą, czyli autodestrukcją komórek, które uczestniczą w rozwoju struktur twarzy. Nie jest jednak do końca jasne w jaki sposób zredukowanie rRNA ogranicza rozwój twarzy.
Penetracja w przypadku dziedziczenia autosomalnego dominującego wynosi 90%. Ekspresja genu jest zmienna nawet w obrębie tej samej rodziny. Z uwagi na zmienną ekspresję, poradnictwo genetyczne jest utrudnione.
Objawy i diagnostyka
Cechy dysmorficzne dotyczą przede wszystkim głowy i twarzy. Ich nasilenie jest zmienne. U jednych pacjentów zmiany będą dyskretne, a u innych ciężkie. Zaliczamy do nich:
- antymongoidalne ustawienie powiek (skośne oczy) i rozszczep powiek dolnych oczu, a także brak lub rzadkie rzęsy na rozszczepionej powiece, przysrodkowo od rozszczepienia,
- niedorozwój ściany bocznej i dna oczodołu,
- niedorozwój kości jarzmowej, widoczny jako brak policzków,
- szpiczasty nos i zarośnięcie nozdrzy tylnych,
- niedorozwój kości jarzmowej, widoczny jako brak policzków,
- niedorozwój małżowin usznych (wystające uszy) i ucha wewnętrznego, niedosłuch przewodzeniowy (zarośnięcie kanału słuchowego, patologie kosteczek słuchowych i ucha środkowego),
- gęstsze owłosienie w okolicach baków,
- makrostomia - przerost jednej strony ust,
- rozszczep podniebienia, podniebienie gotyckie - wąskie i wysoko wysklepione,
- hipoplazja żuchwy - mała, cofnięta bródka (retrognacja, mikrognacja),
- trójkątna twarz, "smutny wyraz twarzy".
 |
| Cięzkie dysmorfie u dziewczynki z zespołem Treacher Collinsa http://www.documentingreality.com |
Rozwój ruchowy i umysłowy dziecka jest normalny. Niestety w codziennym funkcjonowaniu często pojawiają się trudności. Dotyczą one głównie problemów z oddychaniem i karmieniem (z powodu mikrognacji), oczami np. suche oczy i duże ryzyko infekcji oczu (z powodu rozszczepu powiek czy też ich niedomykania) oraz utrudnione chwytanie z powodu małych kciuków.
Stwierdzając zespół Treachera Collinsa, konieczne jest zaobserwowanie wyżej wymienionych objawów, a także wykonanie diagnostyki obrazowa (zdjęcia rentgenowskie, rezonans magnetyczny i tomografia komputerowa głowy). Dziecko wymaga konsultacji okulistycznej, chirurgicznej, plastycznej, chirurga szczękowego, laryngologicznej i innych w zależności od potrzeb. Stale powinno się znajdować pod opieką neonatologa, później pediatry oraz genetyka. Po urodzeniu dziecka możliwe są komplikacje oddechowe z uwagi na cofniętą żuchwę. Intubacja w sytuacji zagrożenia życia może być utrudniona. Potwierdzeniem diagnozy jest wykrycie mutacji patogennej w badaniach genetycznych.
Możliwe jest przeprowadzenie diagnostyki prenatalnej. W rutynowym badaniu USG można stwierdzić wady twarzoczaszki jak np. mikrognacja.
 |
| https://www.sonoworld.com |
Wady w postaci skośnych szpar powiekowych, niedorozwoju policzków i mikrognację, lepiej widać w USG 3D:
 |
https://www.sonoworld.com
|
Jeśli znana jest mutacja wywołująca zespół Treachera Collinsa w danej rodzinie, możliwe jest wykonanie inwazyjnej diagnostyki prenatalnej. W tym celu konieczne jest przeprowadzenie amniopunkcji lub biopsja kosmówki i analiza genetyczna pobranego materiału.
W diagnostyce różnicowej należy wziąć pod uwagę:
- zespół Nagera >>
- zespół Miller’a,
- inne dyzostozy kończynowo-twarzowe,
- połowiczą makrosomię twarzy,
- zespół Goldenhar'a >>
- embriopatia retinoidową.
Możliwości terapeutyczne
Nie ma możliwości przyczynowego leczenia zespołu Treachera Collinsa. Dziecko z tym zespołem wymaga
stałej opieki rodzica, a także częstych konsultacji pediatrycznych. Jeśli wystąpi poporodowa niewydolność oddechowa, może być konieczne zaintubowanie dziecka lub wykonanie tracheostomii i wspomaganie oddechu przez tlenoterapię lub wentylację respiratorem.
W zależności od stopnia zniekształceń twarzy proponowane jest leczenie rekonstrukcyjne kości:
- rekonstrukcja ściany bocznej i dna oczodołu,
- rekonstrukcja kości jarzmowych (6-7 r.ż.),
- rekonstrukcja szczęki i podbródka (14–16 r.ż.).
Ponadto wykonywane są operacje plastyczne powiek dolnych, rozszczepu powiek
(2-3. r.ż.), nosa (14–16 r.ż.) oraz uszu (8–14 r.ż.). W wieku kilku miesięcy -
a przed ukończeniem 2 r.ż., powinno być skorygowane rozszczepione
podniebienie.
Plastyka makrostomii jest zaopatrywana przed ukończeniem pierwszego
roku życia. Zabiegi ortognatyczne - czyli w obrębie szczęki i zębów dopiero po
ukończeniu 14–16 r.ż.
Takie wieloetapowe leczeniem pozwala uniknąć nadmiernej
liczby operacji. Korekcje plastyczne są wykonywane wtedy, gdy wzrost dziecka
już zostaje fizjologicznie zaniechany. W przypadku niedosłuchu, poleca się
założenie aparatu słuchowego.
Przy wdrożeniu odpowiedniego leczenia, rokowanie dla postaci łagodnych zespołu Treachera Collinsa jest dobre.
Na podstawie: dostępy z dn .13.01.2018
- GHR: https://ghr.nlm.nih.gov/condition/treacher-collins-syndrome#definition
- ORPHANET: http://www.orpha.net/consor/cgi-bin/OC_Exp.php?Lng=GB&Expert=861
- NORD: https://rarediseases.org/rare-diseases/treacher-collins-syndrome/
- FACES: http://www.faces-cranio.org/Disord/Treacher.htm
- WSPÓLNIE: http://www.wspolnie.org/zespoly-genetyczne-przewodniki/8-zespoly-genetyczne/70-treacher-collins
- SONOWORLD: https://www.sonoworld.com/Fetus/page.aspx?id=2564



Jaki może być stopień ubytku słuchu u takich dzieci?
OdpowiedzUsuń