Zespół Bardeta-Biedla

- BBS, ang. Bardet-Biedl Syndrome - zespół Bardeta-Biedla
- LMBBS, ang. Laurence-Moon-Bardet-Biedl Syndrome - zespół Laurencea-Moona-Bardeta-Biedla
- zespoły związane z genami BBS - ang. BBS-realted syndrome
| 110 | Q87.8 |
Rozpowszechnienie:
Oszacowano, że w Ameryce Północnej i Europie, zespół Bardeta-Biedla dotyczy od 1 : 140 000 do 1 : 160 000 noworodków. Zauważono, że BBS jest częściej notowany w populacji Nowofundlandzkiej (u wschodniego wybrzeża Kanady), gdzie dotyka około 1 na 17 000 noworodków oraz beduińskiej populacji Kuwejtu: około 1 na 13 500 noworodków.
Przyczyny i dziedziczenie:
W roku 2000 odkryto pierwszą mutację powodującą zespół Bardeta-Biedla. Dotychczas opisano ich 26 i określane są jako "geny BBS". Białka produkowane przez te geny biorą udział w utrzymaniu i funkcjonowaniu rzęsek.
Gdy w genie BBS dojdzie do mutacji, wówczas obserwowane są zmiany w strukturze i czynności rzęsek. Defekty te najprawdopodobniej zaburzają przekazywanie istotnych sygnałów chemicznych podczas rozwoju i prowadzą do nieprawidłowości w percepcji sensorycznej. Uważa się, że wadliwe rzęski są odpowiedzialne za większość cech zespołu Bardeta-Biedla.
Około 1/4 wszystkich przypadków zespołu Bardeta-Biedla jest związana z mutacjami genie BBS1, a kolejna 1/5 jest spowodowana mutacjami w genie BBS10. Pozostałe geny BBS odpowiadają za niewielki odsetek wszystkich przypadków tego zespołu. Warto zauważyć, że w przypadku 1/4 pacjentów przyczyna zespołu Bardeta-Biedla pozostaje nieznana.
Mutacje BBS dziedziczone są w sposób autosomalne recesywny, co oznacza, że dziecko chore dziedziczy od każdego rodzica po jednej zmienionej kopii genu BBS na chromosomach innych niż chromosomy płci. W przypadku nosicielstwa BBS u obojga rodziców, istnieje statystyczne 25% ryzyko na posiadanie z zespołem BBS, 50% na przekazanie bezobjawowego nosicielstwa oraz 25% szans na posiadanie zdrowego dziecka. Ryzyka dla każdych ciąż są takie same.
Objawy i diagnostyka
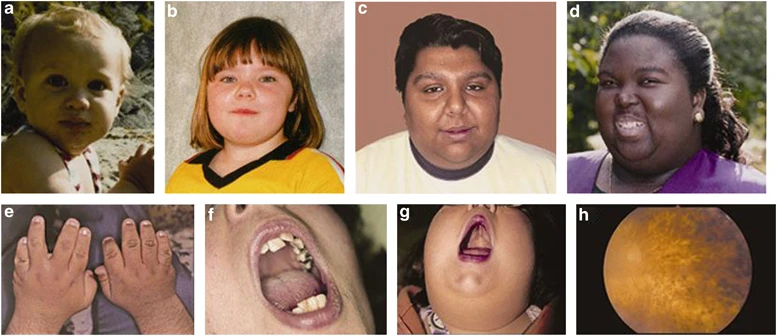
Objawy składające się na zespół Bardeta-Biedla są różnorodne i zmienne, nawet w obrębie jednej rodziny. Wynika to z obecności rzęsek w wielu typach komórek ludzkiego organizmu. Do głównych cech zespołu Bardeta-Biedla zaliczamy: dystrofią czopkowo-pręcikową siatkówki, otyłość i powiązane z nią powikłania (niealkoholowe stłuszczenie wątroby, cukrzyca, nadciśnienie tętnicze), polidaktylia (dodatkowe palce), osłabienie funkcji poznawczych, hipogonadyzm hipogonadotropowy (zaburzenie polegające na niewystarczającym wydzielaniu hormonów gonadotropowych, co prowadzi do niewydolności jąder u mężczyzn oraz jajników u kobiet; niedoboru hormonów płciowych, zaburzenia dojrzewania i wystąpienia niepłodności) i/lub wadami układu moczowo-płciowego oraz wadami nerek i/lub chorobą miąższu nerek.
Pacjenci z zespołem BBS mogą również wykazywać inne nieprawidłowości związane z oczami, jak zez, astygmatyzm i zaćma. Nieco rzadziej obserwuje się choroby przewodu pokarmowego i wątroby, brachydaktylię (krótkie palców), syndaktylię (zrośnięcie palców), nieprawidłowości układu mięśniowo-szkieletowego, zaburzenia neurorozwojowe, w tym łagodną hipertonię, ataksję (zaburzenia koordynacji ruchowej), problemy z zachowaniem równowagi ciała, opóźnienie rozwoju, napady padaczkowe, zaburzenia rozwoju mowy (odbiorczej i ekspresywnej, wady artykulacyjne oraz nosowa i/lub "powietrzna" mowa), utrata słuchu, anosmię (brak węchu) lub osłabienie węchu, nieprawidłowości w obrębie jamy ustnej i zębów (stłoczenie zębów, hipodoncja, wysoko wysklepione podniebienie). Ponadto obserwuje się zaburzenia behawioralne i psychiatryczne, w tym zachowania obsesyjno-kompulsywne, lęk i zaburzenia nastroju oraz niedojrzałość emocjonalną.
Obecne są również delikatne cechy dysmorficzne twarzoczaszki, ale nie stanowią one istotnego kryterium diagnostycznego:
- brachycefalia (spłaszczenie i poszerzenie czaszki w części potylicznej),
- makrocefalia (nieprawidłowo duża czaszka)
- wąskie czoło (skrócona odległość między skroniami),
- krótkie, wąskie i skośne szpary powiekowe,
- głęboko osadzone i szeroko rozstawione oczy (hiperteloryzm),
- duże uszy,
- długa i gładka rynienka podnosowa,
- obniżony grzbiet nosa,
- spłaszczenie kości jarzmowej,
- retrognacja (cofnięta żuchwa).
Wiek
|
Kryteria podstawowe BBS |
Kryteria dodatkowe |
Wskazówki i wymogi diagnostyczne |
Okres prenatalny | - Polidaktylia (obecność palców dodatkowych) - Hipechogeniczne nerki (uszkodzenie/choroba nerek) | - Hydrometrocolops (puchlina pochwy i macicy) - Odwrócenie trzewi (narządów wewnętrznych) | Rozpoznanie pewne: - dodatni wynik badania genetycznego płodu - przynajmniej jedno kryterium podstawowe Rozpoznanie o umiarkowanej pewności: - dodatni wynik BBS u rodzeństwa - przynajmniej jedno kryterium podstawowe lub: dwa kryteria główne i jedno dodatkowe (wskazana pilna diagnostyka genetyczna płodu) |
Od urodzenia do 16. roku życia | - Polidaktylia - Wczesnodziecięca otyłość - Dystrofia siatkówki o wczesnym początku - Anomalie/dysfunkcje nerek | - Hydrometrocolops - Mikropenis (wada rozwojowa polegająca na występowaniu prącia o nieprawidłowo małym rozmiarze) - Zaburzenia neurorozwojowe - Anosmia/hiponosmia (zaburzenie zmysłu węchu polegające na jego braku lub wyraźnym osłabieniu) | Rozpoznanie pewne: - dodatni wynik badania genetycznego dziecka - przynajmniej jedno kryterium podstawowe lub: jeśli diagnostyka genetyczna nie jest możliwa: przynajmniej cztery podstawowe kryteria lub: jeśli diagnostyka genetyczna nie jest możliwa: przynajmniej trzy kryteria podstawowe i dwa dodatkowe Rozpoznanie o umiarkowanej pewności: jeśli diagnostyka genetyczna nie jest możliwa: - dodatni wynik badania genetycznego BBS u rodzeństwa - co najmniej dwa kryteria podstawowe |
Powyżej 16. roku życia i w wieku dorosłym | - Polidaktylia - Wczesnodziecięca otyłość - Dystrofia siatkówki o wczesnym początku - Anomalie/dysfunkcje nerek | - Hipogonadyzm - Mikropenis - Niepełnosprawność neurorozwojowa - Anosmia/hiponosmia | Rozpoznanie pewne: - dodatni wynik BBS - zwyrodnienie siatkówki - co najmniej jedno kryterium podstawowe lub: jeśli diagnostyka genetyczna nie jest możliwa: - zwyrodnienie siatkówki - co najmniej trzy kryteria podstawowe lub: jeśli diagnostyka genetyczna nie jest możliwa: - zwyrodnienie siatkówki - co najmniej dwa kryteria podstawowe - co najmniej dwa kryteria dodatkowe Rozpoznanie o umiarkowanej pewności: jeśli diagnostyka genetyczna nie jest możliwa u pacjenta: - rodzeństwo z BBS (dodatni wynik u rodzeństwa) - co najmniej dwa kryteria główne |
Podsumowując, BBS należy podejrzewać jeśli u płodu lub noworodka/niemowlęcia stwierdzono w obrazie ultrasonograficznym strukturalną chorobę nerek, wady układu moczowo-płciowego i/lub palce dodatkowe, ponieważ mogą to być jedyne objawy widoczne w tych okresach rozwoju.
Diagnostyka różnicowa
Możliwości leczenia
Z uwagi na etiologię zespołu Bardeta-Biedla, jest to zespół niewyleczalny, ale możliwe jest wdrażanie leczenia objawowego i poprawiającego funkcjonowanie pacjentów. Istotne są regularne kontrole stanu zdrowia. W piśmiennictwie znajdują się następujące propozycje:
- ogólnorozwojowe: (antropometria, styl życia: żywienie i aktywność fizyczna) - przy każdej wizycie u lekarza.
- oczy i wzrok: pierwsza konsultacja okulistyczna u niemowląt i małych dzieci oraz dorosłych - następnie co rok lub według zaleceń okulisty.
- jama ustna i zęby: rutynowa opieka stomatologiczna; co pół roku od pierwszego roku życia.
- wady sercowo-naczyniowe oraz ocena klatki piersiowej i jamy brzusznej: ECHO serca w razie występowania wad serca czy kardiomiopatii; USG trzewi z ich oceną i położeniem; w razie anomalii dalsze kontrole zależne od decyzji lekarza prowadzącego.
- zaburzenia ze strony układu oddechowego, przewodu pokarmowego - co rok
- choroba wątroby: gdy brak - coroczna kontrola; w razie choroby - pozostawanie w regularnej kontroli u hepatologa
- choroba nerek: gdy brak - coroczna kontrola; w razie choroby - pozostawanie w regularnej kontroli u nefrologa
- wady urogenitalne: kontrola urologiczna co rok
- zespół metaboliczny (ocena lipidogramu i hemoglobiny glikowanej): co rok od 4. roku życia jeśli wyniki w normie; w razie choroby - częste kontrole zależne od decyzji lekarza prowadzącego (internista, diabetolog)
- niedoczynność tarczycy (co rok)
- hipogonadyzm sipogonadotropowy (USG narządów układu rozrodczego, badanie poziomu hormonów płciowych w razie opóźnienia dojrzewania): co rok począwszy od wieku 13 lat, jeśli jest wskazane (endokrynolog dziecięcy)
- wady mięśniowo-szkieletowe (konsultacja ortopedyczna w razie potrzeby, jeśli występują objawy skoliozy, polidaktylii lub choroby stawów)
- rozwój psychoruchowy (ocena rozwoju i zdolności kognitywnych; rozważenie diagnostyki obrazowej rezonansu magnetycznego jeśli występują objawy neurologiczne) - rutynowe badania bilansowe
- ocena psychiatryczna/behawioralna (ocena neuropsychiatryczna jeśli występują objawy nietypowych zachowań czy zaburzeń nastroju): w razie potrzeby (należy zwrócić uwagę na fakt, że postępująca utrata wzroku i potęgująca się niepełnosprawność oraz zależność w okresie adolescencji i wczesnej dorosłości jest dramatem dla młodego człowieka, toteż wymagają oni szczególnej uwagi i troski bliskich osób)
- konsultacja genetyczna (omówienie wyniku badania genetycznego i implikacji rodzinnych oraz medycznych)
- wsparcie bliskich osób (zaproponowanie sorzystania z zasobów mediów społecznościowych jak „Rodzic dla Rodzica”, pomoc w zorganizowaniu wsparcia socjalnego i psychologicznego dla rodziny
Interwencje terapeutyczne podejmowane są w zależności od indywidualnych potrzeb, czyli objawów, które dominują funkcjonowanie pacjenta. Pacjent powinien zostać objęty opieką wielodyscyplinarnego zespołu lekarzy i terapeutów.
- Dystrofia czopkowo-pręcikowa: z powodu nieuchronnej utraty wzroku niezbędne jest planowanie edukacji; kluczowe jest nauczenie się korzystania z alfabetu Braille'a, trening mobilności i orientacji przestrzennej z białą laską; adaptacji do życia oraz obsługi komputera/smartfona (w tym oprogramowania do rozpoznawania mowy i transkrypcji), a także korzystanie z przyrządów (np lupa cyfrowa) do czytania w dużym druku, gdy wzrok staje się słabszy i szczątkowy; warto rozważyć edukację w ośrodku szkolnym dla dzieci i młodzieży słabowidzących i niewidomych.
- Otyłość: dieta niskokaloryczna, zróżnicowana z ograniczeniem spożycia węglowodanów prostych oraz regularne ćwiczenia aerobowe, takie jak spacery, wędrówki piesze, jazda na rowerze i pływanie z adaptacjami dla osób niewidomych; pomocne okazuje się także poradnictwo dietetyczne oraz fizjoterapia;
- Zespół metaboliczny i inne powikłania BBS związane z otyłością np. cukrzyca i nadciśnienie tętnicze: leczyć w oparciu o standardowe i aktualne wytyczne.
- Anosmia/hiposmia: stosowanie alternatywnych sposobów wykrywania niebezpiecznych substancji (np. zepsutego jedzenia, dymu, gazu), poprzez czujniki czy życzliwość innej osoby.
- Choroby nerek, układu pokarmowego i wątroby, niedoczynność tarczycy i hipogonadyzm należy leczyć tak, jak w populacji ogólnej.
- Wady anatomiczne, w tym polidaktylia, wady uzębienia i zgryzu, wrodzone wady serca, wady układu moczowo-płciowego i nieprawidłowości układu mięśniowo-szkieletowego, mogą wymagać interwencji chirurgicznej.
- W przypadku opóźnienia rozwoju i/lub zaburzeń funkcji poznawczych postępowanie powinno być dostosowane indywidualnie do wieku oraz zidentyfikowanych potrzeb.
- Indywidualna terapia ABA dla dzieci ze spektrum autyzmu - jest ukierunkowana na mocne i słabe strony dziecka w zakresie zachowania, umiejętności społecznych i adaptacji.
Opracowano na podstawie:
- MedlinePlus: https://medlineplus.gov/genetics/condition/bardet-biedl-syndrome/
- Forsyth R, Gunay-Aygun M Bardet-Biedl Syndrome Overview. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJ, Gripp KW, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 2003 [cited 2022 Feb 9]. Available from: http://www.ncbi.nlm.nih.gov/books/NBK1363/ Dostęp z dn. 17.12.2025
- Kerr EN, Bhan A, Héon E. Exploration of the cognitive, adaptive and behavioral functioning of patients affected with Bardet-Biedl syndrome. Clin Genet. 2016 Apr;89(4):426-433. doi: 10.1111/cge.12614. Epub 2015 Jun 16. PMID: 25988237.
- Behavioural phenotype of Bardet-Biedl syndrome
- Dostęp z dn. 17.12.2025



Komentarze
Prześlij komentarz