Zespół Angelamana - "dzieci marionetki"
Harry Angelman, to lekarz, który po raz pierwszy opisał przypadek trojga dzieci z charakterystycznym zestawem objawów. Zainspirowany obrazem Chłopiec z Marionetką, Giovanniego Franceski Caroto, z uwagi na zaobserwowane odmienności u dzieci, takich jak dziwaczne ruchy i "uśmiechnięte" twarze, zatytułował swój raport: Dzieci marionetki. Sztuka bywa więc natchnieniem dla medycyny...
Ritratto di fanciullo con disegno Giovanni Francesco Caroto - Wikipedia |
Zespół Angelmana to rzadka choroba genetyczna, którą cechują zmiany w funkcjonowaniu układu nerwowego, niepełnosprawność umysłowa niecodzienny sposób poruszania się, a także dysmorfie twarzy. Przebieg ciąży jest prawidłowy, a dzieci rodzą się dobrze rozwinięte. Mają właściwą masę ciała i odpowiednią jego długość. Pierwsze niepokojące objawy, związane z zaburzeniami rozwoju psychomotorycznego, obserwuje się między 6., a 9., sporadycznie do 12. miesiąca życia. Dzieci mają poważne trudności z mową. Wymagają też stałej opieki drugiej osoby.
Inne nazwy choroby:
- ang. Angelman syndrome, AS.
Numery w klasyfikacjach:
OMIM | ORPHA | ICD-10 |
| 105830 | Q93.5 |
Częstość występowania: 1/10000 do 1/20000 urodzeń na całym świecie.
Przyczyny i dziedziczenie
Objawy, z jakimi się spotykamy w przypadku zespołu Angelmana są wywołane błędnym funkcjonowaniem genu UBE3A na 15. chromosomie, w miejscu 15q11-q13. Koduje on enzym znany jako ligaza ubikwityny E3A. Jest ona enzymem niszczącym zdegradowane białka w komórkach. Enzymy te przywiązują małe białko, zwane ubikwityną do białek, które powinny być zniszczone. Struktury komórkowe określane jako proteasomy, rozpoznają i trawią oznaczone ubikwityną białka. Proces degradaciji białek w komórce jet potrzebny, ponieważ usówa on zbędne i uszkodzone białka, co zapewnia prawidłowe funkcjonowanie komórek. Badania sugerują, że ligaza ubikwityny odgrywa dużą rolę w prawidłowym rozwoju i funkcjonowaniu układu nerwowego. Jednak jej rola w komórkach układu nerwowego, nie jest do końca jasna.
Ludzie zwykle dziedziczą dwie kopie genu UBE3A, po jednym od każdego z rodziców. Obie kopie tego genu są włączone, czyli aktywne, w większości tkanek organizmu. W niektórych obszarach mózgu, jednak tylko kopia odziedziczona od matki (kopia matczyna) jest aktywna. Ta specyficzna rodzicielska aktywacja genu zależy od niżej opisanego zjawiska imprintingu genomowego.
Zazwyczaj więc mamy do czynienia z tzw. preferencyjną ekspresją allelu matczynego w mózgu, co oznacza, że wada pochodzi od matki.Błędy, zakłócenia w funkcjonowaniu genu UBE3A mogą wynikać z delecji fragmentu chromosomu 15 w obrębie genu, która jest pochodzenia matczynego (gdy jest pochodzenia ojcowskiego, wówczas ujawni się zespół Pradera-Willego). Delecja to innymi słowy wypadniecie fragmentu chromosomu. Kolejną przyczyną zakłóceń w genie UBE3A może być disomia uniparentalna lub
jednorodzielska (UDP). Jest to zjawisko, polegające na odziedziczeniu od jednego rodzica dwóch kopii wadliwych genów - w tym przypadku po ojcu. Taka mutacja zwykle daje łagodniejszy przebieg choroby, z mniejszą ilością napadów padaczkowych, a stanowi ona 1-2% wszystkich przypadków AS. Następnym czynnikiem zakłócającym opisywany gen jest defekt
imprintingu - ID (naznaczenie genetyczne, zaburzenie do którego dochodzi już podczas wytwarzania komórek rozrodczych). Obserwowany jest w około 3%
przypadków. Wreszcie gen UBE3A nie funkcjonuje prawidłowo w przypadku różnych mutacji w tym genie, co spotykane jest w kilku-kilkunastu przypadkach.
Zespół rzadko kiedy jest dziedziczony. Zwykle są to przypadkowe mutacje. AS może czasem wystąpić u następnego dziecka, co dzieje się w przypadku UPD lub nowej mutacji. Takie ryzyko wynosi mniej niż 1%. Podwyższone ryzyko urodzenia dziecka z zespołem Angelmana obserwowane jest u kobiet, będących nosicielkami aberracji zrównoważonej w regionie 15q11-q13 chromosomu.Aberracja to rodzaj mutacji, polegający na zmianie struktury lub liczby - w tym przypadku chromosomów.
Objawy i diagnostyka
W obrazie klinicznym AS, najczęściej występują zaburzenia neurologiczne. Zaliczamy do nich niezborność ruchową, czyli ataksję, napady epileptyczne (padaczka) oraz upośledzenie umysłowe. Dziecko porusza się w specyficzny sposób - chodzi jak marynarz po statku (chód marynarski, na szerokiej podstawie). Miewa też dziwne napady śmiechu.
Okres noworodkowy
Brak objawów. Dzieci rodzą się zwykle o czasie, mają prawidłową masę ciała - czasem kilkaset gramów mniejszą od normy. Długość ciała, obwody główki i klatki piersiowej także są w normie.
Niemowlęctwo
Dzieci mają trudności z ssaniem piersi. Występują ulewania i wymioty po posiłku - często przez nos. Niemowlę może krztusić się mlekiem, co też bywa przyczyną infekcji układu oddechowego. Ciało dziecka jest wiotkie, słabo napięte, lekko drży. Niemowlak sprawia wrażenie spokojnego, cichego, niesprawiającego problemów. Około szóstego miesiąca życia pojawiają się pierwsze objawy zaburzeń rozwojowych, jak spowolnienie rozwoju psychoruchowego, jednak AS jest diagnozowany dopiero w wieku przedszkolnym - od 2. do 5. roku życia, kiedy to objawy zespołu są najpełniej wyrażone.
Okres poniemowlęcy i dziecięcy
Obserwuje się w nim opóźnienie rozwoju, nabywania umiejętności mowy oraz zmiany w zachowaniu dziecka. Zaburzenia rozwoju psychoruchowego wynikają z wiotkości mięśni, zwłaszcza tułowia i nadmiernego napięcia mięśni w kończynach. Dziecku drżą nogi, jego ruchy są niezgrabne, nieskoordynowane. Nie trzyma ono równowagi i przejawia ruchy mimowolne, niejednokrotnie szybkie. Dzieci śpią krócej niż ich zdrowi rówieśnicy.
Siad dziecka - zwykle w wieku około 1 do 3 lat, jest specyficzny: wiotkość tułowia sprawia, iż dziecko się podpiera jedną ręką, a jego nogi są sztywne i szeroko rozstawione.
Około 1,5 roku można zaobserwować raczkowanie podobne do czołgania się, a także przesuwanie się na pośladkach po podłożu. Dzieci źle się czują w pozycji na brzuchu, ale ta je aktywizuje do wstawania. Chodzić zaczynają w wieku od 1,5 roku do 7 lat. Chód jest niepewny, chwiejny, na szerokiej postawie. Nogi dziecka są sztywne, z powodu wysokiego napięcia mięśni.
Jeżeli dziecko ma skrzywienie kręgosłupa, czyli skoliozę, bardzo nasiloną wiotkość mięśni korpusu oraz wysokie napięcie mięśni kończyn, wówczas może nie chodzić. Dzieje się tak u około 1/3 chorych dzieci.Jeżeli dzieci nie są rehabilitowane - grożą im przykurcze kolanowe i progres skoliozy.
Jeśli chodzi o sposób bycia dziecka i jego zachowania - często jest ono obierane jako pogodne, wesołe i towarzyskie. Miewa napady śmiechu. Śmiech dziecka pojawia się nieadekwatnie do sytuacji, łatwo go wywołać, ale nie można go opanować i kontrolować. Nie jest to jednak kryterium zespołu, pomimo wysokiej częstości występowania.
Dzieci fascynują się bardzo otoczeniem: wodą, światełkami, karbowanym i kolorowym papierem, celofanami i różnymi dźwiękami. Dobrze czują się w kąpieli i bawiąc się zabawkami, które wydają odgłosy. Cieszą je "świetlne zajączki", odgłos odkurzacza i puszczona woda z kranu.
Charakterystycznym objawem AS, są zaburzenia mowy. Często są to poważne ubytki. Dzieci wypowiadają pojedyncze słowa lub nie mówią wcale. Mają jednak dobrze opanowane alternatywne sposoby komunikacji np. gestami, lekkim pociąganiem za rękę opiekuna lub jego ubranie.
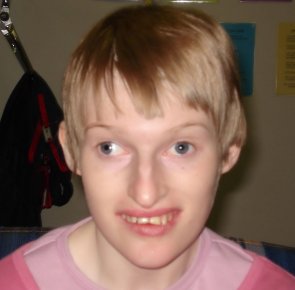
Wygląd dziecka
Głowa jest często mała i krótka (małogłowie i krótkogłowie), potylica głowy nierzadko płaska. Podstawa czaszki jest stromo ustawiona. W okresie noworodkowym nie widać żadnych zmian, dopiero później obserwuje się dysmorfie określane mianem "twarzy Angelmana". Do jej cech zaliczamy:
 |
| http://www.primehealthchannel.com |
- ciągłe wysuwanie języka, najpierw między zęby, potem między usta, a później nawet na brodę,
- postępujący prognatyzm (silne wysunięcie szczęk do przodu), mała żuchwa, później bródka wydatna, a dolna część twarzy poszerzona,
- cechy hipoplazji (niecałkowite wykształcenie) w centralnej części twarzy,
- głęboko osadzone oczy, hiperterolyzm oczny, czasem zmarszczki nakątne,
- delikatne rysy twarzy,
Uzębienie dzieci jest szeroko rozstawione, a zęby mają starte powierzchnie, z uwagi na stale wysuwający się język. "Twarz Angelmana" nie przysłania podobieństwa do rodziców. Dzieci mają jasne włosy i skórę. Jest ona wrażliwa na promienie słoneczne. W tęczówkach stwierdza się niedobór barwnika. Takie zjawisko nosi nazwę hipopigmentacji. Ponadto u dzieci może wystąpić zez i krótkowzroczność.
Dorośli z AS
Problemy zdrowotne dorosłych z zespołem Angelmana są podobne jak w przypadku dzieci, czyli dotyczą: skolioz, przykurczy, padaczki i zachowania. Nie tracą oni nabytych umiejętności, ale wymagają opieki do końca życia. Niekiedy wskazane są u nich leki neuroleptyczne i uspokajające, jeśli chodzi o zaburzenia behawioralne i bezsenność.
Długość życia chorych z zespołem Angelmana może wynosić nawet 70 lat, ale nie ma doniesień na ten temat.
Objawy wynikające z zakłóceń genu UBE3A
- delecje: małogłowie, napady padaczkowe, hipopigmentacja, nieprawidłowy ruch, zaburzenia mowy i myślenia,
- disomia uniparentalna lub
jednorodzielska i defekt imprintingu: mniej dymorfizmu, napadów padaczkowych, zaburzeń ruchu,
- defekt imprintingu: łagodniejsze zaburzenia mowy, zdolności poznawczych i ruchowe,
- mutacje UBE3A: objawy umiarkowane jeśli chodzi o małogłowie, napady padaczkowe, zaburzenia ruchu i mowy - wszsytko zależy od jakości mutacji.
Badania genetyczne
W wielu laboratoriach w Polsce wykonywane są badania molekularne zespołu Angelmana. Są to zwykle:
- analiza chromosomowa w poszukiwaniu dużych delecji oraz mikrodelecji (FISH),
- analiza metylacji w badanym regionie DNA,
- analiza polimorfizmu markerów mikrosatelitarnych,
- poszukiwanie mutacji w genie UBE3A.
Różnicowanie z:
- zespołem Lennoxa-Gastauta,
- zespołem Pitta-Hopkinsa,
- zespołem Retta,
- zespołem Mowat-Wilsona,
- autyzmem niemowlęcym.
Diagnostyka prenatalna
Jest wykonywana jeśli są ku temu istotne wskazania. Zwykle wykonuje się badania inwazyjne, jak biopsja kosmówki, czy amniopunkcja, z badaniami genetycznymi. Wykorzystywane są techniki FISH i analiza metylacji DNA.
Zalecane kontrole i konsultacje:
- neurologiczna (padaczka, ataksja, spastyka),
- okulistyczna (zez, krótkowzroczność),
- psychiatryczna (zaburzenia zachowania, upośledzenie umysłowe),
- gastroenterologiczna (ulewania, wymioty),
- ortopedyczna (skoliozy, przykurcze),
- logopedyczna (ćwiczenia aparatu mowy, nauka komunikacji werbalnej i niewerbalnej),
- w gabinecie stomatologicznym,
- psychologiczno-pedagogiczna.
Możliwości terapeutyczne
W rehabilitacji ruchowej polecana jest metoda NDT-Bobath i ćwiczenia w wodzie (które zwłaszcza ulubiły sobie dzieci z zespołem Angelmana). Pozwalają one spowolnić progres skoliozy i przykurczów, a także poprawić równowagę i koordynację ruchową.
Terapia pedagogiczna powinna obejmować integrację sensoryczną, Metodę Ruchu Rozwijającego Weroniki Sherborne, dogoterapię i hipoterapię, a także Program Aktywności Knill, Metodę Felicie Affolteri, malowanie dziesięcioma palcami.
Ulewania w okresie niemowlęcym i ewentualny refluks żołądkowo-przełykowy, można łagodzić poprzez leczenie dietetyczne. Najlepszym pokarmem okażą się gęste mieszanki i mleka modyfikowane dla dzieci, z symbolem AR (antyrefluks).
Istotna jest także nauka komunikacji pozawerbalnej, jak np. piktogramy, gesty, mimika oraz system komunikacji symbolicznej Bliss.
Czy wiesz, że...
Synek Collina Farrela - James jest dzieckiem zmagającym się z zespołem Angelmana. Początkowo stwierdzono u niego mózgowe porażenie dziecięce, ale później zweryfikowano diagnozę.
 |
| http://www.celebritydiagnosis.com |
Colin Farrell opowiada o zespole Angelmana u Ellen
Warto zajrzeć:



{kind=link}
{kind=link}
Oby jak najmniej dzieci dotykały takie i podobne przypadłości.:)
OdpowiedzUsuńNikt nie chce być chory i nikt nikomu nie życzy źle :) Przynajmniej w moim otoczeniu...
OdpowiedzUsuńprawdopodobnie moja coreczka ma zespol angelmana
OdpowiedzUsuńMoja córcia ma Zespół Angelmana.
OdpowiedzUsuńNigdy nie słyszałam o takiej chorobie. Ciekawy artykuł:)
OdpowiedzUsuńMoja siostrzyczka na ten zespół... :( dowiedzieliśmy sie o tym wczoraj :( ;(
OdpowiedzUsuńDziś odebralismy wynik - diagnoza Zespół Angelmana :((
OdpowiedzUsuńDostaliśmy wyniki w środę. Synek ma oprócz autyzmu zespół Angelmana.
OdpowiedzUsuńJestesmy w trakcie badan.Wszystkie objawy tu przeczytane to tak jak u mojej corci.
OdpowiedzUsuń