Choroba Gauchera - choroba spichrzeniowa
Choroba Gauchera to genetycznie uwarunkowane zaburzenie metaboliczne,
dzidziczone w sposób autosomalny recesywny. Mutacje w genie GBA (gen kodujący
enzymu o nazwie beta-glukocerebrozydaza, który rozkłada tłuszczową substancję
glukocerebrozydu do cukru (glukoza) oraz prostsze cząsteczki tłuszczu
(ceramidu)). W wyniku defektu enzymu dochodzi do aktywnego gromadzenia
nieprawidłowych lipidów w śledzionie, wątrobie i szpiku kostnym (komórki
Gauchera).
Inne nazwy choroby: Acid beta-glucosidase deficiency,
Glucocerebrosidase deficiency
Rozpowszechnienie: 1-9:100 000. Częstość występowania GD w populacji
ogólnej wynosi około 1/60, 000, ale może wynieść do 1/1, 000 w populacji Żydów
aszkenazyjskich.
Przyczyny i
dziedziczenie
Choroba dziedziczona autosomalnie recesywnie. Główną rolę odgrywa mutacja w
genie GBA, kodującego lizosomalny enzym o nazwie beta-glukocerebrozydaza.
Rozkłada on glukocerebrozyd (substancja tłuszczowa), do glukozy (czyli cukru)
oraz ceramidu (prostsze cząsteczki tłuszczu). W wyniku mutacji i związanego z
nią brakiem aktywności enzymu, dochodzi do gromadzenia się (spichrzania)
nieprawidłowych tłuszczów w wątrobie, śledzionie i szpiku kostnym, co prowadzi
do powiększenia i niszczenia tych narządów i upośledzenia ich funkcji.
W bardzo rzadkich przypadkach, mutacja występuje w genie PSAP, kodującego
białko aktywujące glukocerebrozydazę – sapozynę C (saposin C)
Objawy i przebieg
Nagromadzenie się
lipidów prowadzi do znacznego powiększenia śledziony, śledziony i nieprawidłowego modelowania kości na tzw. kształt kolby Erlenmeyera, osteopenii, z czego wynika zwiększone ryzyko patologicznych złamań, w tym kompresyjnych złamań kręgów
(miażdżenie kręgów, charakterystyczne dla osteoporozy), co daje objawy w późnym dzieciństwie lub na początku okresu dojrzewania
(10-14 lat). Objawy choroby są bardzo zmienne. W zależności od zaobserwowanych
objawów klinicznych, wyróżniamy trzy typy choroby:
TYP I (nieneuropatyczny) – najczęściej ośrodkowy
układ nerwowy (mózg i rdzeń) nie są zajęte. Może przebiegać w postaci łagodnej,
umiarkowanej lub ciężkiej. To nie-neurologiczna forma związana z organomegalią
(powiększenie śledziony i wątroby), anomaliami kości (ból, martwica kości,
złamania patologiczne) i niedoborem krwinek. Objawy pojawiają się w każdym
momencie życia – od dzieciństwa po dorosłość. Najbardziej dotkliwe w wieku dorosłym są dolegliwości kostne.
Pojawiają się przewlekłe,
dokuczliwe bóle, niekiedy przyjmują postać ostrych kryz bólowych, opornych na
standardowe leczenie przeciwbólowe. Zajęcie układu kostnego dobrze widać w MRI (rezonans
magnetyczny) jako nacieczenie szpiku kostnego makrofagami i zmianę proporcji frakcji tłuszczowej szpiku. W badaniach krwi stwierdza się zwykle: niedokrwistość i małopłytkowość, często uogólnioną neutropenię oraz
zaburzenia krzepnięcia, wysoką ferrytynę i niski poziom
witaminy B12. Typ I dotyczy 90% przypadków.
Warto zapamiętać!
U dzieci dochodzi do spowolnienia lub zahamowania tempa
wzrastania oraz opóźnienie dojrzewania.
Objawy występujące prawie zawsze u dorosłych z typem I choroby
Gauchera to:
- uczucie zmęczenia, osłabienie, bóle kostne,
- wzmożona tendencja do krwawień (krwawienia z nosa, dziąseł, wylewy podskórne, hypermenorrhoea),
- powiększenie śledziony i wątroby (znaczne rozmiary nawet kilkudziesięciokrotne)
Często występują:
- zawały śledziony,
- patologiczne złamania, martwica kości (najczęściej stawów
biodrowych),
- niedokrwistość, małopłytkowość.
Rzadziej obserwuje się:
- nadciśnienie płucne,
- marskość wątroby.
TYP II (neuropatyczny) – cechą charakterystyczną jest zajęcie
OUN (oczopląs, drgawki, uszkodzenie mózgu), to ostra forma dziecięca choroby
Gauchera, która najczęściej powoduje ciężkie zmiany i śmierć w okresie
dziecięcym (około 2. roku życia). Dysfunkcja pnia mózgu szybko postępuje, a
choroba jest także związana z organomegalią. Obserwuje się objawy opuszkowe i spastyczność.
TYP III (neuropatyczny) – również
charakteryzuje się uszkodzeniem OUN, ale może postępować wolniej niż typ II. To
podostra
neurologiczne forma dotykająca dzieci lub młodzieży, a cechuje ją postępująca
encefalopatia (apraksja gałek ocznych w ruchach horyzontalnych, ataksja, zez,
mioklonie, niewielkie upośledzenie umysłowe) z
ogólnoustrojowymi objawami obserwowanymi w typie I. Forma płodowa przejawia się
ze zmniejszeniem lub brakiem ruchów płodu lub puchliną (anasarca, obrzęk).
Choroba
podobna do choroby Gauchera, objawia się stopniowym zwapnienia aorty i zastawki
aortalnej i/lub mitralnej - będącymi jej głównymi cechami.
Diagnostyka
Oficjalne rozpoznanie
choroby zależy od pomiaru poziomu glukocerebrozydazy w krążących leukocytach
lub hodowlanych fibroblastów skóry. Diagnozę potwierdza genotypowanie.
Różnicowanie
Diagnostyka
różnicowa obejmuje inne lizosomalne choroby spichrzeniowe. Obecność komórek
podobnych do komórek Gauchera można znaleźć w przypadku niektórych chorób
hematologicznych, jak chłoniak, chłoniak Hodgkina i przewlekła białaczka
limfocytowa.
Możliwości terapeutyczne
Dostępne są
dwa sposoby leczenia typu GD 1 i 3: enzymatyczna terapia zastępcza (stosowanie imiglucerazy
lub welaglucerazy) i terapia redukcji substratów (miglustat). Opisywane metody
nie są skuteczne dla typu GD 2.
Rokowanie jest dobre w rodzaju GD 1. W typie 2, zgon pojawia się zwykle przed ukończeniem 2 roku życia. Bez szczególnego postępowania, typ GD 3 prowadzi do śmierci w ciągu kilku lat.
Rokowanie jest dobre w rodzaju GD 1. W typie 2, zgon pojawia się zwykle przed ukończeniem 2 roku życia. Bez szczególnego postępowania, typ GD 3 prowadzi do śmierci w ciągu kilku lat.
Na podstawie:
Praca poglądowa: Choroba Gauchera - Anna Tylki-Szymańska
Choroba Gauchera - Dr hab. n. med. Tomasz Kulpa
Historia zdjęcia...
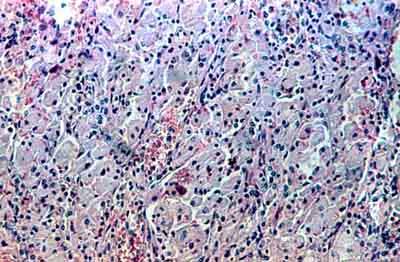
... Tym razem to nie ja wykonała to jakże inspirujące zdjęcie. Znalazłam je na portalu Medycyny Praktycznej mp.pl. Jak niektórzy zdążyli się domyślić, to obraz z badania bioptycznego histopatologicznego. Mp.pl podaje: "Na zdjęciu widoczne znacznie powiększone komórki Browicza i Kupffera oraz histiocyty przestrzeni wrotnych obładowane glukocerebrozydami (komórki o cytoplazmie wyglądającej jak zmięty papier)"... Choć ja tu widzę mak rozsypany we fiolecie ;)
***
Historia zdjęcia...
... Tym razem to nie ja wykonała to jakże inspirujące zdjęcie. Znalazłam je na portalu Medycyny Praktycznej mp.pl. Jak niektórzy zdążyli się domyślić, to obraz z badania bioptycznego histopatologicznego. Mp.pl podaje: "Na zdjęciu widoczne znacznie powiększone komórki Browicza i Kupffera oraz histiocyty przestrzeni wrotnych obładowane glukocerebrozydami (komórki o cytoplazmie wyglądającej jak zmięty papier)"... Choć ja tu widzę mak rozsypany we fiolecie ;)



Komentarze
Prześlij komentarz