Dysplazja kampomeliczna
 Dysplazja kampomeliczna jest bardzo rzadko występującą wrodzoną dysplazją kostną. Dysplazja to zaburzenie rozwojowe polegające na nieprawidłowej organizacji i funkcjonowaniu komórek w określonej tkance. Dysplazję kampomeliczną charakteryzuje współwystępowanie różnie nasilonych wad układu szkieletowego, jak wygięcie i kruchość kości długich, anomalie klatki piersiowej i miednicy z niejednoznacznymi cechami płciowymi lub odwróceniem płci. Z greki kampomelia oznacza - "wygiętą kończynę".
Dysplazja kampomeliczna jest bardzo rzadko występującą wrodzoną dysplazją kostną. Dysplazja to zaburzenie rozwojowe polegające na nieprawidłowej organizacji i funkcjonowaniu komórek w określonej tkance. Dysplazję kampomeliczną charakteryzuje współwystępowanie różnie nasilonych wad układu szkieletowego, jak wygięcie i kruchość kości długich, anomalie klatki piersiowej i miednicy z niejednoznacznymi cechami płciowymi lub odwróceniem płci. Z greki kampomelia oznacza - "wygiętą kończynę".Synonimy:
- Acampomelic campomelic "Dysplasia" - akampomeliczna kampomeliczna dysplazja (jeśli nie stwierdza się wygięcia kości długich),
- Campomelic Dwarfism, CMDI - niskorosłość kampomeliczna,
- Campomelic Dysplasia - dysplazja kampomeliczna,
- Campomelic Syndrome, Long-Limb Type - zespół kampomeliczny, typu długich kończyn
- Camptomelic Dwarfism - niskorosłość kamptomeliczna,
- Camptomelic Syndrome - zespół kamptomeliczny,
- Camptomelic Syndrome, Long-Limb Type - zespół kamptomeliczny, typu długich kończyn
- SRY-Box 9, SOX9 Mutations Syndrome - zespół mutacji SRY-Box 9, SOX9.
Rozpowszechnienie: konkretnie nieokreślone. Oszacowano je na 1:40 000-1:200 000 osób. Uważa się, że dysplazja kampomeliczna dwa razy częściej dotyczy kobiet niż mężczyzn - jednak dane mogą być błędne, z uwagi na interpretację niejednoznacznych cech płciowych lub rewersję płci. Do roku 2007 opisano około 100 przypadków.
Przyczyny i dziedziczenie
Dysplazja kampomeliczna wynika z mutacji w genie SOX9 na chromosomie 17. Około 5% przypadków jest spowodowanych zaburzeniami chromosomowymi, które występują w pobliżu genu SOX9 - ich przebieg jest zazwyczaj łagodniejszy niż powodowanych przez mutacje w genie SOX9.
Mutacja dziedziczona jest w sposób autosomalny dominujący. Może pojawiać się de novo, jak również występować rodzinnie (np. wśród rodzeństwa). Gen SOX9 koduje białko zawierające instrukcje dla prawidłowego tworzenia się chrząstek i kształtowania układu płciowego już w okresie embrionalnym.
Objawy i diagnostyka
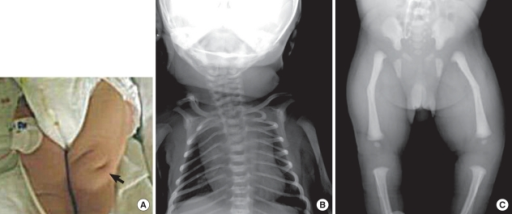 |
A - dołki skórne; B - klatka dzwonowata, hipoplastyczna czaszka
i łagodna skolioza piersiowo-lędźwiowa; C - wygięcie kości długich.
Kim HY et all. (2010)
|
Postawienie diagnozy możliwe jest już w okresie prenatalnym podczas wykonywania USG w drugim trymestrze ciąży. Można wtedy dostrzec wielowodzie, opóźniony rozwój związany z wadami kostnymi, nieproporcjonalnie dużą głowę, krótkie kończyny, a także męski pseudohermafrodytyzm. Jeśli znana jest mutacja powodująca chorobę, wówczas możliwe jest wykonanie amniopunkcji genetycznej lub biopsji kosmówki.
Noworodki z dysplazją kampomeliczną najczęściej umierają niedługo po urodzeniu z powodu
niewydolności oddechowej. Warto mieć na uwadze, że około 5-10% dzieci przeżywa dłużej, zatem nie wolno z góry skazywać ich na przegraną.
Do charakterystycznych cech dysplazji kampomelicznej zaliczamy:
- wygięcie kości udowych i piszczelowych (na zdjęciach rentgenowskich można też dostrzec, że kości są cienkie), nad miejscami maksymalnego wygięcia kończyn obserwuje się dołki skórne,
- podłużna i wąska czaszka,
- wydatne (wypukłe) czoło, "płaska twarz", wystające oczy, obniżona nasada nosa, mała żuchwa i krótka szyja.
Czasami dochodzi do przedwczesnego zamknięcia szwów czaszkowych i wówczas czaszka dziecka ma kształt trójlistnej koniczyny, co określamy trifoliocefalią. Osoby z dysplazją kampomeliczną miewają też cechy zespołu Pierre'a Robina (rozszczep podniebienia, glosoptoza - język opadający, rozwinięty bardziej z tyłu niż normalnie; i mikrognacja - nieprawidłowo mała żuchwa).
Klatka piersiowa jest wąska i dzwonowata. Stwierdza się jedenaście par żeber zamiast dwunastu. Ponadto występują słabo rozwinięte łopatki i wady stóp, jak stopa końsko-szpotawa.
Razem z wadami układu szkieletowego, często występują anomalie układu płciowego (choć nie są konieczne do stwierdzenia dysplazji kampomelicznej). Większość pacjentów z kariotypem męskim prezentuje fenotyp płci żeńskiej, co określane jest jako rewersja płci (zespół odwrócenia płci). Wówczas zewnętrzne narządy płciowe nie odpowiadają wewnętrznym. U chorych stwierdza się zaburzenia rozwoju gonad i większą tendencję do nowotworów narządów rodnych.
Wraz z wiekiem u pacjentów może rozwijać się kifoskolioza, a także ujawniają się wady bioder (małe, dyslokujące się) i niedosłuch. Może dochodzić do ucisku kręgów na rdzeń kręgowy. Osoby z dysplazją kampomeliczną częściej zapadają na infekcje układu oddechowego, które mogą doprowadzać do niewydolności oddechowej i wcześniejszych zgonów (podobnie jak występująca u niektórych osób wąska krtań, hipoplazja pierścieni tchawicy i wiotkość krtani/tchawicy). Czasem obserwowane są trudności w uczeniu się (lekkie do umiarkowanych). Pacjenci nie osiągają prawidłowego wzrostu.
W zdjęciach rentgenowskich stwierdza się:
- znacznie wydłużoną czaszkę z wąskimi, płytkimi oczodołami,
- cienkie żebra, których jest jedenaście par; dzwonowaty kształt klatki piersiowej,
- anomalie kręgów i zmniejszenie odległości między nasadami łuków kręgowych głównie w odcinku lędźwiowym,
- wygięcie kości długich, które są szczupłe,
- wąską, podłużna, hipoplastyczną miednicę.
Różnicowanie:
- wrodzona łamliwość kości,
- achondroplazja,
- hipofosfatazja,
- dysplazja tanatoforyczna,
- hipoplazja chrząstek i włosów,
- nie związane z zespołem genetycznym wygięcie kości.
Możliwości leczenia
Dysplazja kampomeliczna na dzień dzisiejszy pozostaje chorobą niewyleczalną, ale możliwe jest leczenie objawowe. Wady ortopedyczne, takie jak skoliozy, podwichnięcia stawów biodrowych, stopy końsko-szpotawe powinny być korygowane i monitorowane corocznie przez lekarza ortopedę. W razie potrzeby wykonywane są operacje zgodnie z obowiązującymi procedurami.
Pediatra powinien obserwować i dokumentować tempo wzrostu. Osoby z dysplazją kampomeliczną nie osiągają normalnego wzrostu. Należy też ocenić funkcjonowanie układu oddechowego. W przypadku niewydolności oddechowej wdrażana jest nieinwazyjna wentylacja mechaniczna z utrzymaniem dodatniego ciśnienia w drogach oddechowych. Do niewydolności oddechowej dochodzi w wyniku zespołu niewydolności klatki piersiowej. Ten ostatni można próbować korygować operacyjnie poprzez system implantów VEPTR (Vertebral Expandable Prosthetic Titanium Rib). Metoda ta pozwala na poprawę sylwetki pacjenta poprzez zmianę kształtu jego klatki piersiowej, co z kolei daje szansę na poprawę wydolności oddechowej. Zabieg jest bardzo rozległy i polega na założeniu odpowiednich implantów i skorygowaniu deformacji, a następnie zastosowaniu dystrakcji (rozciągania). Jeśli implanty założono u dzieci, wówczas biorąc pod uwagę wzrost układu kostnego dokonuje się rozsuwania implantów co około pół roku. Kolejne zabiegi są mniej inwazyjne i hospitalizacja trwa krócej.
Nieprawidłowe gonady powinny zostać usunięte z uwagi na ryzyko rozwinięcia się nowotworu gonadoblastoma. U pacjentek z zespołem odwrócenia płci istotne jest monitorowanie stężenia alfa-fetoproteiny (AFP) i antygenu rakowo-płodowego (CEA) w celach prognostycznych. Ponadto warto dokonywać regularnych oznaczeń gonadotropin (głównie: folitropiny i lutropiny) oraz estradiolu i testosteronu.
Dla osób z utratą słuchu zaleca się stosowanie aparatów słuchowych.
Rodzinom, w których urodziło się dziecko z dysplazją kampomeliczną wskazana jest konsultacja i poradnictwo genetyczne.
Na podstawie:
- Unger S. Campomelic Dysplasia. Gene Reviews Internet. Initial Posting: July 31, 2008; Last Update: May 9, 2013.
- Petriczko E. Dysplazja kampomeliczna i zespół odwrócenia płci – sześcioletnia obserwacja. Endokrynol. Ped., 12/2013;1(42):77-82
- Genetics Home Reference. Campomelic dysplasia. 2014
- Orphanet. Campomelic dysplasia. 2007
- National Organization for Rare Disorders. Campomelic Syndrome. 2007
- Kim HY, Yoon CH, Kim GH, Yoo HW, Lee BS, Kim KS, Kim EA -A case of campomelic dysplasia without sex reversal. J. Korean Med. Sci. (2010)
- Zarzycki D. et all. Zastosowanie systemu implantów VEPTR (Vertebral Expandable Prosthetic Titanium Rib) w leczeniu wrodzonych deformacji krêgos³upa i klatki piersiowej. Przegląd Lekarski 2008 / 65 / 7-8



Komentarze
Prześlij komentarz