Dysplazje przynasadowe - McKusicka i Schmida
 Kolejnymi chondrodysplazjami które poznamy są dysplazje przynasadowe (chondrodysplasia
metaphysealis). Cechami charakterystycznymi tych chorób są anomalie tzw. płytki wzrostowej, czyli
chrząstki nasadowej. Chrząstka ta znajduje się w pobliżu zakończeń rosnącej
jeszcze kości. To miejsce w którym przyrasta ona na długość. W chrząstce
tej w fazie wzrostu pojawiają się następne punkty kostnienia później przekształcające ją w kość.
Kolejnymi chondrodysplazjami które poznamy są dysplazje przynasadowe (chondrodysplasia
metaphysealis). Cechami charakterystycznymi tych chorób są anomalie tzw. płytki wzrostowej, czyli
chrząstki nasadowej. Chrząstka ta znajduje się w pobliżu zakończeń rosnącej
jeszcze kości. To miejsce w którym przyrasta ona na długość. W chrząstce
tej w fazie wzrostu pojawiają się następne punkty kostnienia później przekształcające ją w kość.
W dysplazji przynasadowej choroba
ujawnia się właśnie w częściach przynasadowych kości długich i krótkich. We wspomnianej
płytce wzrostowej dochodzi do zaburzeń układu chondrocytów (podstawowych komórek tkanki chrzęstnej). Zamiast typowego układu kolumnowego komórek chrząstki, tu obserwuje się
nieregularne skupiska chondrocytów.
Dysplazja przynasadowa występuje w
różnych postaciach. Najczęściej opisywane są: dysplazja przynasadowa McKusicka
(dysplasia cartilagopilaris) i dysplazja przynasadowa typu Schmida.
Dysplazja przynasadowa McKusicka
Dysplazja przynasadowa McKusicka
znana też jako zespół McKusicka, cartilage-hair
hypoplasia (hipoplazja chrząstkowo-włosowa), CHH, autosomal recesive
metaphyseal chondrodysplasia, metaphyseal chondrodysplasia - jest dziedziczona w
sposób autosomalny recesywny. Rozpowszechnienie wady jeszcze nie zostało
oszacowane. Jednak zdecydowanie częściej dotyczy Amiszy i Finów.
Dysplazja McKusicka jest spowodowana przez mutację w genie RMPR na chromosomie 9 (locus 9p21-p12.). Wspomniany gen nie koduje białka, ale komponentę RNA enzymu mitochondrialnej endorybonukleazy RNA. Objawy dysplazji pojawiają się już u noworodków lub niemowląt. Cechy kliniczne wyróżniające ten typ dysplazji to:
Dysplazja McKusicka jest spowodowana przez mutację w genie RMPR na chromosomie 9 (locus 9p21-p12.). Wspomniany gen nie koduje białka, ale komponentę RNA enzymu mitochondrialnej endorybonukleazy RNA. Objawy dysplazji pojawiają się już u noworodków lub niemowląt. Cechy kliniczne wyróżniające ten typ dysplazji to:
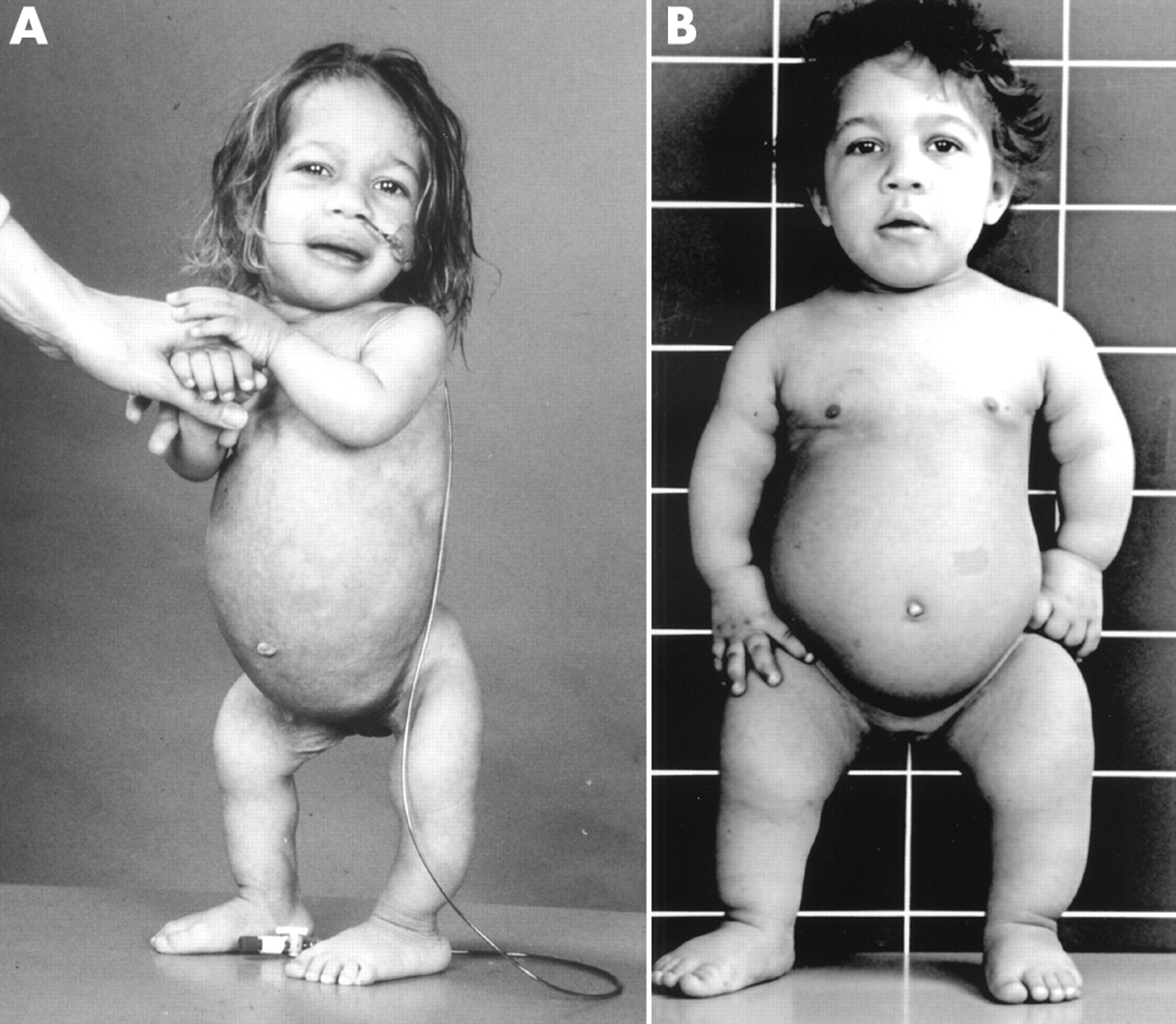 |
| http://jmg.bmj.com/content/40/10/761.full |
-
niski wzrost,
-
krótkie kończyny – w częściach bliższych (mikromelia rizomeliczna)
-
krótkie dłonie z nadmierną ruchomością w stawach,
-
zmiany chorobowe w kręgosłupie, przynasadach kości (przynasady nieregularne,
„postrzępione”) ,
-
skąpe, słabo ubarwione, wolno rosnące owłosienie ciała,
-
zaburzenia odporności (limfopenia – za mało komórek odpornościowych zwanych
leukocytami i defekt odporności komórkowej).
W
radzie podejrzenia dysplazji McKusicka potwierdzeniem jest badanie
sekwencjonowania genu RMRP. Diagnostyka różnicowa powinna obejmować inne
postaci karłowatości z krótkimi kończynami.
Ryzyko pojawienia się dysplazji przynasadowej u kolejnego dziecka wynosi 25%, co uzasadnia diagnostykę prenatalną na podstawie analiz molekularnych (biopsja trofoblastu lub amniopunkcja genetyczna). Jest to możliwe wówczas, gdy u probanta (osobnika, który „zapoczątkował” chorobę) określono jaki ma rodzaj mutacji. Jeśli chodzi o diagnostykę prenatalną nieinwazyjną – możliwe jest zaobserwowanie skrócenia kończyn w badaniu USG, ale nie jest to objaw specyficzny i na 100% potwierdzający diagnozę.
Zaburzenia
odpornościowe – w tym przypadku niedobór odporności może być tak poważny, że wymaga przeszczepu szpiku kostnego, ale nie ma to istotnego wpływu na wyrównanie
niedoboru.
Rokowanie w tej dysplazji zależy od obecności i ciężkości niewydolności układu odpornościowego i możliwego związku z chorobą Hirschsprunga.
Rokowanie w tej dysplazji zależy od obecności i ciężkości niewydolności układu odpornościowego i możliwego związku z chorobą Hirschsprunga.
Dysplazja przynasadowa typy Schmida
Dysplazja przynasadowa typu
Schmida jest rzadkim schorzeniem dziedziczonym w sposób
autosomalny dominujący.
Spowodowana jest przez mutacje genu COL10A1 na chromosomie 6 (locus 6q21-q22).
Gen COL10A koduje łańcuch kolagenu alfa-1(X). Rozpowszechnienie dysplazji
przynasadowej jest jeszcze nieznane.
 |
| http://caseconnector.jbjs.org |
Chorobę charakteryzuje
umiarkowanie niski wzrost, czyli karłowatość, krótkie kończyny, biodro szpotawe
(vara coxa), pałąkowate – szpotawe kończyny dolne (bowlegs) oraz zaburzenia
chodu. Dysplazja przynasadowa jest zwykle rozpoznawana w drugim lub trzecim
roku życia.
Diagnoza opiera się na wykryciu
zmian w częściach przynasadowych kości podczas radiografii (zdjęcia rentgenowskie). Diagnostyka różnicowa
powinna obejmować:
- hipochondroplazję,
- następstwa krzywicy (niedobór
witaminy D).
Pozostałe dysplazje przynasadowe
(np. opisana wyżej dysplazja McKusicka i dysplazja przynasadowa typu Jansen (Jansen type metaphyseal chondrodysplasia))
mogą zostać wykluczone z różnicowania, ponieważ na ich fenotyp składa się bardzo niski wzrost
i inne cechy.
W przypadku stwierdzenia dysplazji Schmida zalecane jest poradnictwo
genetyczne, ponieważ ryzyko powtórzenia się wady wynosi 50%. Wskazana jest
także diagnostyka prenatalna. Jedyne możliwe opcje leczenia to korekcja
ortopedyczna.
Dysplazja przynasadowa typu Jansen
zostanie opracowana w kolejnym wpisie, który już niebawem się ukaże na blogu...



Moja córka ma dysplazję przynasadową McKusica właśnie... Gdy miała 3 miesiące przeprowadzono u niej przeszczep szpiku kostnego, ponieważ jej odporność była bliska zeru... Teraz jest już prawie 5 lat po przeszczepie i niedoboru odporności nie ma w ogóle, nie choruje praktycznie wcale, a poziom białych krwinek jest taki jak u innych zdrowych dzieci. :)
OdpowiedzUsuń